
1 引言:分子定量的黄金标准
在现代分子生物学、医学诊断及生物技术领域,实时荧光定量聚合酶链式反应(Quantitative Real-time Polymerase Chain Reaction,简称qPCR)已确立了其作为核酸定量分析“黄金标准”的地位。从基础的基因表达差异分析到临床病原体载量检测,再到转基因作物鉴定与单核苷酸多态性(SNP)分型,qPCR凭借其极高的灵敏度、宽广的动态范围(可覆盖7至9个数量级)以及精准的定量能力,成为了生命科学实验室不可或缺的核心技术。传统的PCR技术(终点法PCR)主要依赖凝胶电泳在反应结束后对扩增产物进行检测。这种方法虽然能回答“有没有”的问题,但在回答“有多少”时却显得力不从心。原因在于PCR反应遵循指数扩增规律,随着循环数的增加,反应体系中的底物(引物、dNTPs)逐渐耗尽,酶活性降低,反应产物积累导致的抑制作用增强,扩增曲线最终进入“平台期”。在平台期,产物的最终积累量与初始模板量之间不再保持线性关系,因此终点检测无法准确反映样本间的初始差异。
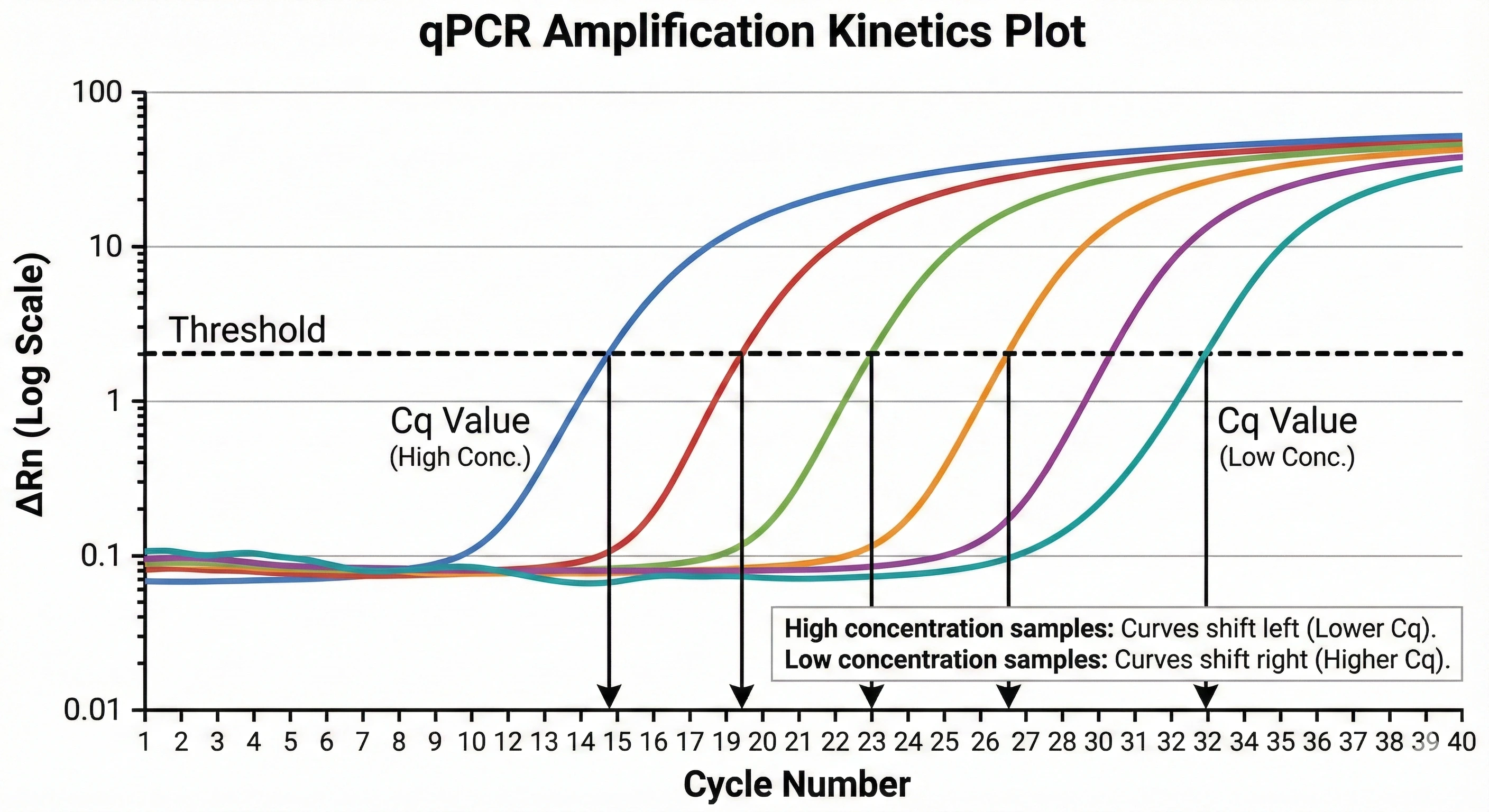
qPCR技术的革命性突破在于引入了荧光化学监测系统。通过在反应体系中加入荧光染料或探针,仪器能够在每一个热循环(Cycle)的延伸阶段实时采集荧光信号。这一技术使得研究者能够观测到PCR反应从背景基线到指数扩增期的完整动力学过程。在指数扩增阶段,荧光信号的增强与扩增产物的增加严格成正比,而达到某一特定荧光阈值所需的循环数(Cq值)与初始模板量的对数成线性反比关系。这种“实时”监测机制巧妙地避开了平台期效应,实现了对初始模板的精确绝对或相对定量 。2 核心原理与荧光化学机制
2.1 反应动力学参数解析
在深入实验之前,必须对描述qPCR扩增曲线的关键术语有深刻的物理与生物学理解:
- 基线(Baseline) :在反应的最初数个循环(通常为第3-15个循环),荧光信号主要由背景噪声(如游离荧光染料的微弱发光、检测器的电子噪声)构成,未见明显的产物信号积累。准确扣除基线是数据分析的第一步。
- 阈值(Threshold) :这是一个人为设定或由软件自动计算的荧光强度值,通常设定在基线噪声标准差的10倍以上,且必须位于对数扩增期的线性区域内。
- 定量循环数(Quantification Cycle, Cq) :旧称Ct(Threshold Cycle)或Cp(Crossing Point)。这是qPCR中最核心的定量参数,指荧光信号强度穿过设定的阈值线时所对应的循环数。Cq值越小,意味着样本中初始靶标模板的拷贝数越高。MIQE指南强烈建议统一使用术语Cq以规范学术交流。
- 扩增效率(Amplification Efficiency, E) :在理想的化学计量条件下,PCR产物在每个循环翻倍,即E=100%(或E=2)。然而,现实中由于引物二聚体干扰、酶活性衰减、抑制剂存在等因素,效率往往在90%-110%之间波动。E值的准确测定对于后续使用Pfaffl法进行数据校正至关重要。
2.2 荧光检测系统:化学原理与选择策略
qPCR的检测灵敏度与特异性在很大程度上取决于所选用的荧光化学系统。目前主流技术分为非特异性嵌合染料法(如SYBR Green)和序列特异性探针法(如TaqMan)。
2.2.1 SYBR Green I 嵌合染料法
SYBR Green I 是一种不对称的花菁染料,具有结合双链DNA(dsDNA)小沟的特性。在游离状态下,其分子结构中的两个芳香环可以自由旋转,通过非辐射跃迁耗散能量,荧光量子产率极低。一旦嵌入双链DNA,分子旋转受阻,荧光量子产率瞬间提升约1000倍。
- 随着PCR扩增的进行,双链产物不断积累,结合的染料分子增多,荧光信号随之增强。
- 优势:成本低廉,通用性强(无需针对每个基因合成探针),且具有独特的功能——熔解曲线分析(Melt Curve Analysis) 。反应结束后,通过程序升温监测荧光信号的骤降,可以根据产物的熔解温度(Tm)区分特异性扩增产物和引物二聚体,这是质量控制的关键手段。
- 局限与挑战:SYBR Green "不挑食",它会结合反应体系中所有的双链DNA,包括非特异性扩增产物和引物二聚体。因此,如果引物设计不佳,可能会产生虚假的荧光信号,导致定量结果偏高。这对引物的特异性设计提出了极高要求。
2.2.2 TaqMan 水解探针法
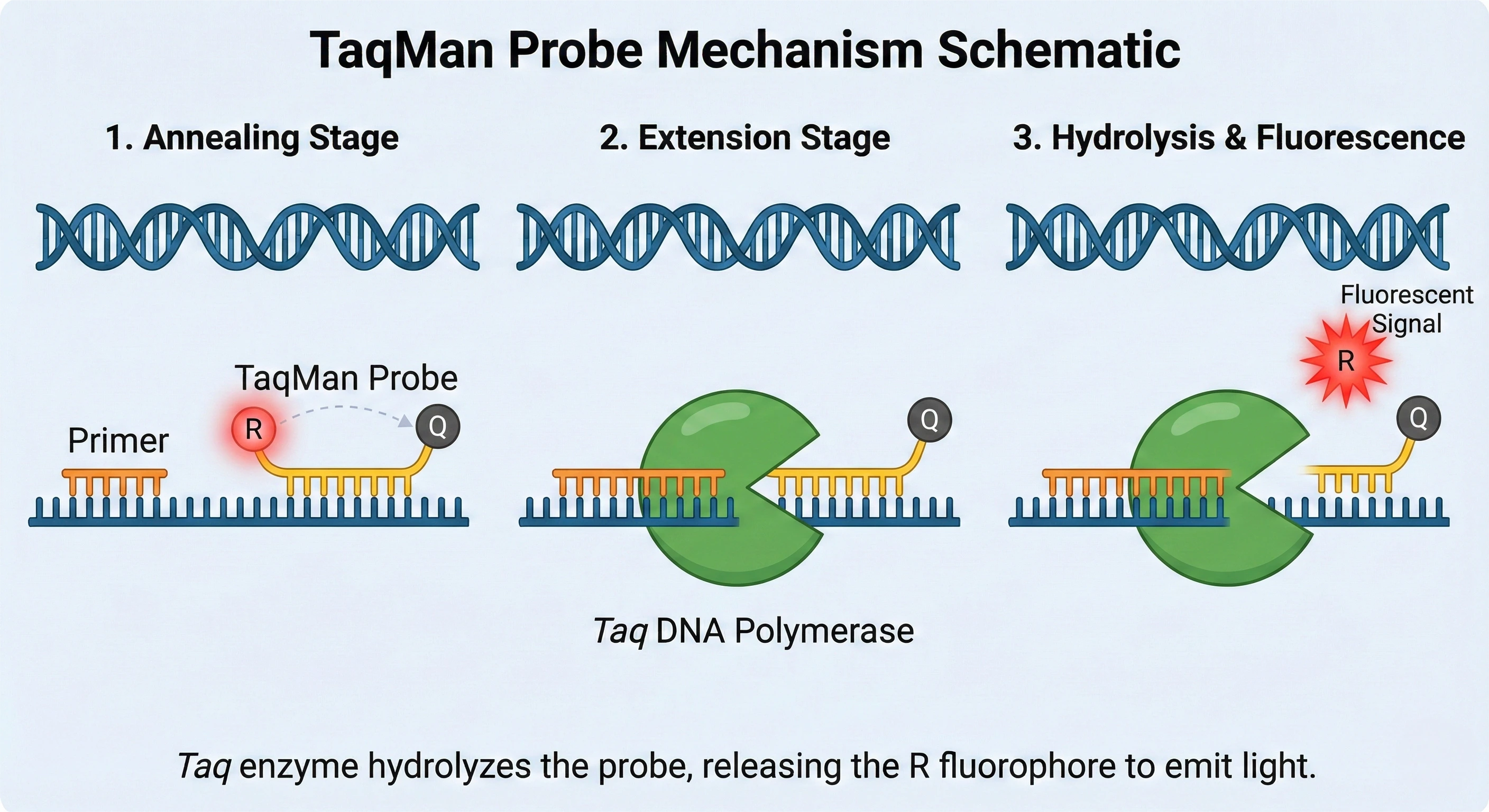
TaqMan系统利用了Taq DNA聚合酶的5'→3'外切酶活性。反应体系中除了引物外,还包含一条特异性结合在引物扩增区域内部的探针。探针的5'端标记有高能量的荧光报告基团(Reporter,如FAM, VIC),3'端标记有淬灭基团(Quencher,如TAMRA, NFQ)。
- 当探针完整时,由于荧光共振能量转移(FRET)效应,报告基团发射的荧光被淬灭基团吸收。在PCR延伸阶段,当Taq酶合成新链遇到探针时,其外切酶活性将探针水解切断,使报告基团游离于淬灭基团之外,从而发出被检测到的荧光。
- 优势:极高的特异性——只有引物和探针同时特异性结合,才能产生信号。此外,通过标记不同颜色的荧光基团,TaqMan技术支持在同一反应管中检测多个靶标(多重PCR,Multiplexing),极大提高了通量并降低了样本消耗。
- 局限:合成探针成本较高,且无法像SYBR Green那样通过熔解曲线排查引物二聚体(引物二聚体不会引起探针水解,因此不发光,但也消耗了底物)。
表 1:SYBR Green 与 TaqMan 化学系统的多维度对比
| 比较维度 | SYBR Green I | TaqMan Probes |
|---|---|---|
| 特异性来源 | 仅依赖引物 | 引物 + 探针双重验证 |
| 荧光信号归属 | 所有双链DNA(含非特异产物) | 仅特异性水解产物 |
| 实验设计复杂度 | 低(仅需设计引物) | 中高(需设计引物和探针) |
| 成本效益 | 初始成本低,适合大规模筛选 | 初始成本高,适合精准定量 |
| 多重PCR能力 | 无(无法区分不同产物) | 有(利用多色荧光通道) |
| 后处理分析 | 必须进行熔解曲线分析 | 无需熔解曲线 |
| 主要应用场景 | 基因表达初筛、验证引物性能 | 临床诊断、SNP分型、低丰度检测 |
3 实验标准化:MIQE指南深度解读
为了解决qPCR领域长期存在的实验设计不严谨、数据报告不透明导致的不可重复性问题,Bustin等国际专家于2009年发布了《定量实时PCR实验出版最低信息指南》(MIQE Guidelines)。这不仅是发表高水平论文的门槛,更是保证实验科学性的基石。3.1 核心检查清单(Checklist)与实践意义
MIQE将报告要素分为“必需”(Essential)和“期望”(Desirable)。忽视这些要素可能导致审稿人拒稿,更严重的是导致错误的生物学结论。
-
实验设计(Experimental Design) :
- 定义实验组与对照组:必须明确样本分组逻辑。
- 生物学重复(Biological Replicates) :指不同来源的独立样本(如不同的小鼠、不同的细胞培养孔)。MIQE要求明确重复数量,这是统计学显著性的基础。
- 技术重复(Technical Replicates) :同一核酸样本在PCR板上的重复孔,主要用于评估移液误差和仪器稳定性。
-
样本制备与质控(Sample Preparation) :
- 核酸完整性:必须报告RNA的质量指标,如RIN值(RNA Integrity Number)或凝胶电泳图。降解的RNA会导致Cq值滞后,严重扭曲定量结果。
- 纯度检测:A260/A280和A260/A230比值是评估蛋白、酚、盐残留的关键。
- 无逆转录对照(No-RT Control) :必需项。用于检测是否存在基因组DNA(gDNA)残留。如果No-RT对照出现扩增,说明RNA提取不纯,必须进行DNase处理。
-
引物与探针信息:
- 序列公开:必须提供引物和探针的核苷酸序列。对于商业化试剂盒,如果序列保密,必须提供Amplicion Context Sequence或准确的定位信息,否则数据可信度存疑 6。
- 特异性验证:需提供BLAST比对结果或凝胶电泳图,证明产物单一。
-
数据分析参数:
- 效率校正:必须报告PCR扩增效率(E值)。盲目假设E=100%是导致相对定量误差的主要来源。
- 归一化方法:详述参比基因(内参)的选择依据及其稳定性验证结果。
3.2 参比基因(内参)的选择策略:超越GAPDH
相对定量的核心是将目标基因表达量与内参基因进行比对,以消除样本投入量、逆转录效率及移液误差的影响。然而,不存在“万能”的内参。传统内参如GAPDH、ACTB在缺氧、细胞凋亡或特定药物处理下,其表达量可能剧烈波动,导致归一化失败 21。专家建议的筛选流程:
-
候选基因库构建:根据物种和组织类型选择候选基因。
- 人/小鼠常用:GAPDH, ACTB, 18S rRNA (丰度极高,需稀释), HPRT1, TBP, YWHAZ, B2M, PPIA, RPLP0。
- 植物常用:UBQ, EF1a, SAND, TIP41。
-
稳定性评估算法:利用geNorm、NormFinder或BestKeeper软件对候选内参在具体实验条件下的稳定性进行排序。
- geNorm:计算基因表达稳定性值(M值),M < 0.5 通常被视为理想。该算法还引入了配对变异分析(V值),用于确定最少需要几个内参才能达到准确的归一化效果。
-
多内参归一化:MIQE强烈建议使用至少2个、最好3个经过验证的内参基因的几何平均数进行归一化,以抵消单个基因的随机波动。
4 实验操作全流程指南
4.1 样本制备:高质量RNA的提取艺术
RNA提取是qPCR成功的起点。由于RNA极易被无处不在的RNase降解,操作过程需严格遵循无RNase原则。目前最经典的方法是异硫氰酸胍-酚-氯仿法(Trizol法)。
Trizol法提取关键步骤深度解析:-
裂解(Lysis) :
- 操作:加入Trizol试剂(含苯酚和异硫氰酸胍),充分匀浆。
- 原理:异硫氰酸胍是强力的蛋白质变性剂,能迅速破坏细胞结构并抑制RNase活性;苯酚则破坏疏水键,使蛋白质变性。此步必须彻底,否则后续分离不纯。
-
相分离(Phase Separation) :
- 操作:加入氯仿,剧烈振荡15秒(呈乳浊状),室温静置2-3分钟,低温离心。
- 原理:氯仿促进有机相(苯酚)与水相的分离。离心后体系分为三层:上层无色水相(含RNA)、中间白色层(变性蛋白和gDNA)、下层红色有机相(蛋白、脂类)。
- 关键点:吸取上清时切勿贪多,宁可丢弃部分水相也不要触及中间层,这是防止gDNA和蛋白污染的最关键步骤。
-
RNA沉淀(Precipitation) :
- 操作:转移上清至新管,加入等体积异丙醇,颠倒混匀,低温沉淀。
- 原理:在水溶液中,核酸表面覆盖水化膜。异丙醇夺取水分子,使RNA分子间因疏水作用聚集沉淀。亦可加入糖原(Glycogen)作为共沉淀剂以提高微量样本的回收率。
-
洗涤(Wash) :
- 操作:弃上清,加入75%乙醇(DEPC水配制),温和颠倒,离心。
- 原理:75%乙醇中的水溶解残留的盐分(如胍盐),而乙醇保持RNA不复溶。
- 关键点:沉淀干燥时间需精准控制(5-10分钟)。过度干燥(沉淀变透明)会导致RNA难以复溶;干燥不足则残留乙醇,强力抑制后续Taq酶活性。
质量控制标准:
- NanoDrop光度法:A260/A280应在1.8-2.1(<1.8提示蛋白/苯酚污染);A260/A230应>2.0(<2.0提示异硫氰酸胍/碳水化合物残留)。
- Agilent Bioanalyzer:测定RIN值。RIN > 7被认为是高质量RNA,适用于qPCR;RIN < 5提示严重降解,建议重新提取。
4.2 逆转录(Reverse Transcription, RT)
将不稳定的RNA转化为稳定的互补DNA(cDNA)。
引物策略选择:- Oligo(dT) :特异性结合mRNA的3'端Poly(A)尾。优点是仅扩增mRNA,背景低;缺点是对5'端序列覆盖度差,且无法扩增无Poly(A)的RNA(如18S rRNA、lncRNA、原核生物mRNA)。
- 随机引物(Random Hexamers/Nonamers) :随机结合RNA各处。优点是覆盖全长转录本,适合降解样本或长基因的5'端检测;缺点是会逆转录所有RNA(包括rRNA),可能导致cDNA产量被rRNA稀释。
- 混合引物(Blended Primers) :多数商业化试剂盒(如Bio-Rad iScript, Thermo SuperScript VILO)采用Oligo(dT)与随机引物的优化配比,兼顾灵敏度与覆盖度,是目前的首选方案。
反应程序:通常包含 25°C(随机引物退火)→ 42-50°C(逆转录酶延伸)→ 85°C(酶失活)。
4.3 反应体系构建与加样技巧
qPCR的灵敏度意味着哪怕是微升(μL)级别的加样误差也会被指数放大。
典型反应体系(20 μL)配:| 组分 | 体积 (μL) | 终浓度 | 作用 |
|---|---|---|---|
| 2X Master Mix | 10.0 | 1X | 含Taq酶、dNTPs、Mg2+、Buffer、SYBR Green |
| 正向引物 (10 μM) | 0.4 - 0.8 | 200-400 nM | 特异性扩增起始 |
| 反向引物 (10 μM) | 0.4 - 0.8 | 200-400 nM | 特异性扩增起始 |
| cDNA模板 | 1.0 - 2.0 | 不定 (<100 ng) | 需预先稀释(如1:5或1:10) 以稀释潜在抑制剂 |
| Nuclease-free Water | 补足至 20 | - | 调整体积 |
| (可选) TaqMan探针 | 0.2 - 0.5 | 100-250 nM | 特异性荧光检测 |
操作黄金法则:
-
配制预混液(Master Mix Premix) :计算所需的总孔数(包含重复和NTC),多配制10%的体积。将除cDNA外的所有试剂预混合,分装后再加入模板。这消除了孔间试剂浓度的差异。
-
物理隔离:在不同的区域进行试剂配制(无DNA区)和加样(DNA区),防止气溶胶污染。
-
设置对照:
- NTC (No Template Control) :用水代替模板,监控试剂污染和引物二聚体。
- IRC (Inter-Run Calibrator) :如果实验跨越多块板,每块板必须包含一个完全相同的阳性对照样本,用于校正板间差异。
5 生物信息学:引物设计实战教程
优秀的引物设计是qPCR成功的一半。设计不当会导致非特异扩增(假阳性)、引物二聚体(消耗底物、低效率)或无法区分gDNA。
5.1 关键设计参数
- 产物长度(Amplicon Size) :70 - 150 bp。短片段扩增效率更高,且受RNA降解影响较小。
- 熔解温度(Tm) :60°C ± 1°C。正反向引物Tm值差异不应超过2°C,以保证同步退火。
- GC含量:40% - 60% 。避免出现长串单一碱基(如GGGG),特别是3'端,以防非特异性粘连。
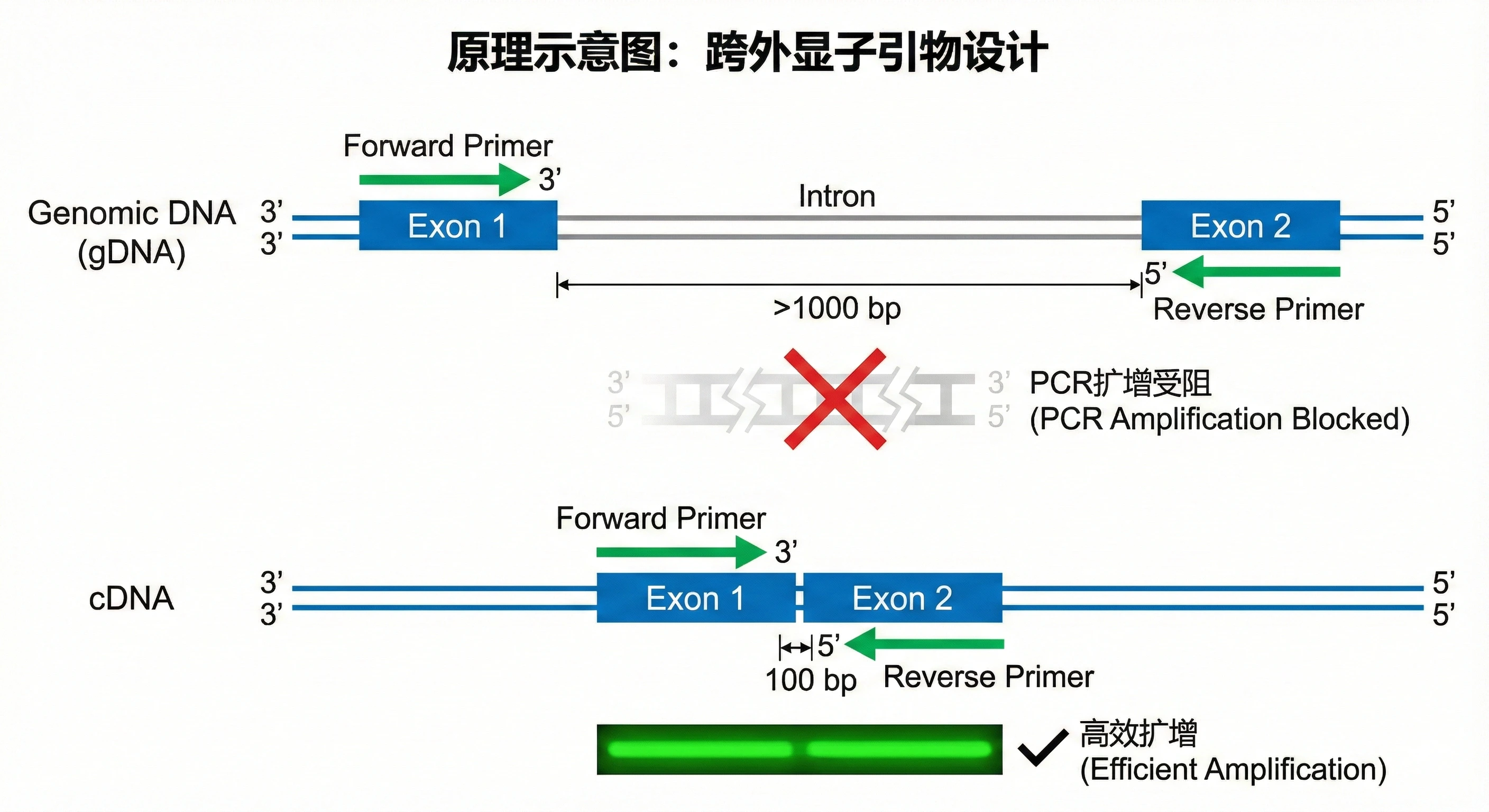
- 跨外显子设计(Exon-Spanning) :核心要求。引物应设计在两个外显子的连接处(Junction),或者正反向引物分别位于被大内含子隔开的两个外显子。
- 原理:cDNA中不含内含子,两引物距离近(~100bp),可有效扩增;gDNA中含内含子,两引物距离远(>1000bp),在qPCR短促的延伸时间(通常30秒)内无法完成扩增,从而避免gDNA信号干扰。
5.2 NCBI Primer-BLAST 操作指南
-
准备序列:在NCBI查找目标基因的RefSeq mRNA序列(如
NM_001234.5)。 -
进入工具:访问 Primer-BLAST 网站。
-
设置参数:
- PCR Template:输入序列号。
- PCR product size:Min
70, Max150。 - Primer melting temperatures:Min
58, Opt60, Max63。 - Exon/Intron Selection:务必勾选 "Primer must span an exon-exon junction" 。
- Organism:选择对应的物种(如 Homo sapiens)。
- Database:选择
RefSeq mRNA以检查转录本特异性,或选择Genome检查全基因组非特异性结合。
-
分析结果:
- 检查引物位置:是否跨越了内含子?
- 检查自互补性(Self-complementarity):分数越低越好,高分意味着易形成二聚体。
- 二次验证:使用IDT OligoAnalyzer等工具再次核算Tm值和二级结构(发卡结构、二聚体自由能\Delta G ), \Delta G应高于 -9 kcal/mol。
6 数据分析:从数学模型到生物学意义
qPCR数据分析不仅仅是读数,它是建立在严格数学假设基础上的计算过程。
6.1 相对定量: 法 (Livak Method)
最常用的基因表达差异分析方法。
公式推导与应用:
假设目标基因(Target)和内参基因(Ref)扩增效率均为100%(E=2)。
-
归一化 (\Delta Cq):消除样本量差异。
\Delta Cq_{\text{Sample}} = Cq_{\text{Target}} - Cq_{\text{Ref}} -
比较 (\Delta\Delta Cq):消除对照组背景。
\Delta\Delta Cq = \Delta Cq_{\text{Treated}} - \Delta Cq_{\text{Control}} -
计算倍数变化 (Fold Change):
\text{Fold Change} = 2^{-\Delta\Delta Cq}
适用条件:目标基因与内参基因的扩增效率必须接近(差异<5%),且都接近100%。需通过标准曲线验证效率一致性。
6.2 效率校正:Pfaffl 方法
当扩增效率不理想或不一致时,Livak法会带来显著误差。此时应使用Pfaffl模型:此处 E 为实际扩增倍数(例如效率95%,则 E=1.95)。
6.3 绝对定量:标准曲线法
用于测定病毒载量或绝对拷贝数。
操作流程:
-
构建标准品:克隆有目标片段的质粒,测定浓度并换算为拷贝数/μL。
\text{Copy Number} = \frac{\text{浓度 (ng/}\mu\text{L)} \times 6.02 \times 10^{23}}{\text{片段长度 (bp)} \times 660 \times 10^9} -
梯度稀释:制备5-7个10倍梯度稀释点(如10^7, 10^6 \dots 10^1)。
-
拟合曲线:以 Log(拷贝数) 为X轴,Cq为Y轴。线性回归方程:Y = \text{Slope} \times X + \text{Intercept}。
-
计算:将未知样本Cq代入方程求X,再取反对数得到拷贝数。
6.4 扩增效率(Efficiency)的计算
通过标准曲线斜率(Slope)计算:
- Slope = -3.32 \rightarrow E = 100\%
- Slope = -3.60 \rightarrow E = 90\% (效率偏低,可能有抑制剂或条件未优化)
- Slope = -3.10 \rightarrow E = 110\% (效率虚高,通常由引物二聚体或非特异扩增引起)
7 故障排除 (Troubleshooting) 深度诊断
qPCR数据异常往往隐含着特定的实验问题。以下是基于症状的深度诊断指南。
7.1 症状一:无扩增信号 (No Amplification) 或 Cq 为 N/A
可能原因与解决方案:
-
表达量过低:靶标丰度低于检测限。
- 诊断:正对照(高表达样本)正常,仅特定样本无信号。
- 对策:增加模板投入量(注意不要引入过多抑制剂);使用巢式PCR;更换检测灵敏度更高的探针法。
-
PCR抑制剂:Trizol残留的苯酚/乙醇或样本中的血红素、多糖抑制Taq酶。
- 诊断:稀释效应测试。将模板进行梯度稀释(如原液, 1:10, 1:100)。如果1:10稀释液的Cq值反而比原液出现得早(或差值远小于3.32),则证实存在抑制剂。
- 对策:纯化模板,或坚持使用稀释后的模板进行实验。
-
引物问题:引物降解或设计失误。
- 诊断:NTC和正对照均无信号。
- 对策:重新合成引物;检查引物Tm值是否高于退火温度。
7.2 症状二:熔解曲线出现双峰或杂峰 (SYBR Green)
图谱解析:
-
引物二聚体 (Primer Dimers) :
- 特征:在较低温度(通常 < 75°C)出现宽峰,且在NTC孔中也存在。
- 原因:引物自身互补,模板浓度过低时引物过剩自连。
- 影响:Cq值被严重高估(假阳性)。
- 对策:优化引物设计(检查3'端互补);提高退火温度;减少引物浓度。
-
非特异性产物:
- 特征:出现多个Tm值较高的峰。
- 原因:扩增了gDNA(含内含子,片段大,Tm高)或非靶标基因。
- 对策:跑胶验证条带大小;重新设计跨外显子引物;进行DNase I 处理。
7.3 症状三:扩增效率异常 (<90% 或 >110%)
-
效率 < 90% (斜率 < -3.6) :
- 原因:PCR抑制剂干扰;引物退火温度过高导致结合不充分;产物长度过长(>200bp)。
- 对策:稀释模板;降低退火温度;重新设计引物。
-
效率 > 110% (斜率 > -3.1) :
- 原因:由于引物二聚体在低浓度样本中占主导。在标准曲线低浓度点,引物二聚体产生荧光,使得Cq值比预期的更早出现,导致斜率变平。
- 对策:这是引物设计不佳的典型标志,必须优化引物。
8 前沿展望:从qPCR到数字PCR (dPCR)
虽然qPCR是当前的主流,但**数字PCR (Digital PCR, dPCR)** 代表了定量的未来方向 。- 原理:dPCR将反应体系分割成数万个微滴(Droplets),每个微滴中包含0或1个模板分子。扩增后,通过统计发光微滴的比例(泊松分布),直接计算绝对拷贝数。
- 对比优势:dPCR不需要标准曲线即可实现绝对定量;对抑制剂的耐受性更强;在检测极低丰度突变(如液体活检中的ctDNA)时灵敏度远超qPCR。
- MIQE升级:针对dPCR,专门发布了dMIQE指南,强调了微滴体积方差、分割数等新参数的报告规范。
9 结论
实时荧光定量PCR是一项将生物学特异性与数学精确性完美结合的技术。获得高质量数据的关键不仅在于昂贵的仪器,更在于研究者对MIQE标准的恪守、对引物设计的严苛以及对每一个Cq值背后动力学含义的深刻理解。通过标准化的实验流程与严谨的数据分析,qPCR将继续作为生命科学探索微观世界的最强有力的量尺。


